

Factores hereditarios
Antitrombina
| Factor Leyden (Resistencia a la proteína C
activada) | Factor II (protrombina) G20210A |
Deficiencias de proteínas C y S | Hiperhomocistinemia
(Homocistinuria) | Disfibrinogenemia |
Alteraciones del sistema fibrinolítico |
Aumento del factor VIII (factor antihemofílivo A)
Cronológicamente, el descubrimiento de una disminución de la actividad de la ATIII representó el primer paso para el reconocimiento de la Trombofilia. Esta glicoproteína es el máximo inhibidor fisiológico de la trombina generada por la cascada de la coagulación. Su efecto enzimático neutralizante también abarca los factores activados IIa (protrombina), Xa, IXa y XIIa (Hageman). En condiciones hemostáticas normales, su inhibición de la trombina es relativamente lenta (actividad inactivante de trombina), pero en presencia de heparina, este proceso se intensifica en una magnitud de 1000-10000 veces (actividad de cofactor de heparina). La presencia de ATIII es indispensable para que, al unirse fuertemente a ella, la heparina pueda ejercer sus efectos. Esta cohesión "obligada" explica la rara observación de una aparente resistencia al efecto heparínico cuando ocurren severas deficiencias o disfunciones de la ATIII.
 La
molécula de ATIII es codificada por medio de 7 exones y 432 codones y
contiene 432 aminoácidos (2). Sus funciones antienzimáticas principales
se concentran alrededor de 2 dominios funcionales:
La
molécula de ATIII es codificada por medio de 7 exones y 432 codones y
contiene 432 aminoácidos (2). Sus funciones antienzimáticas principales
se concentran alrededor de 2 dominios funcionales:
1) Un centro reactivo (arg393 - ser394), que actúa como receptáculo de inserción de la trombina y de los otros factores preactivados. Para el inicio de la actividad anticoagulante, estas enzimas se adosan y escinden enlaces específicos de este centro reactivo, creando compuestos estables enzimáticamente inactivos de la ATIII con cada uno de ellos, los cuales van siendo removidos rápidamente de la circulación, probablemente a través del sistema retículo endotelial.
2) La región captadora de heparina ubicada dentro de 2 áreas contiguas de la zona terminal de la molécula.
Como fenómeno compartido por casi todas las proteínas de la coagulación, los distintos defectos moleculares se manifiestan básicamente por:
A) Una disminución conjunta y proporcional del antígeno y de sus correspondientes funciones (Defecto tipo I)
B) Alteraciones funcionales sin modificaciones de las concentraciones del antígeno (Defecto tipo II).
Por los momentos, y de acuerdo al tipo y localización de las mutaciones, se han identificado 4 alteraciones hereditarias claramente diferenciables (Tabla 3). Para precisarlas se emplean 2 prototipos de ensayos: los funcionales, que evalúan específicamente y por separado la capacidad de interacción con la trombina y la heparina y los inmunológicos, que utilizan anticuerpos purificados anti-ATIII para cuantificar al antígeno (inmunodifusión, método de Laurell, etc.) y analizan las características del patrón de precipitación inmune durante la migración en un campo eléctrico (inmunoelectroforesis cruzada) (5). Las mutaciones correspondientes a cada uno de estos defectos solo pueden verificarse mediante la tecnología de amplificación y partición del ácido desoxirribonucleico (ADN) (6). Para evitar resultados falsamente negativos, todas estas determinaciones deben practicarse a suficiente distancia del empleo de transfusiones sustitutivas.

La incidencia de la deficiencia de ATIII en donantes de sangre es aproximadamente de 1/5000. El defecto tipo I que predispone a complicaciones trombóticas es poco frecuente, al mismo tiempo la aparición de la variedad tipo II heterozigota, caracterizada por un defecto en la captación de heparina y no asociada a riesgo trombótico, se ha estimado en una proporción de hasta 1/700 en individuos normales.
En el manejo de estos defectos no existe una terapia antitrombótica preventiva, debido a que únicamente se logran mantener niveles hemostáticos de ATIII mediante el suministro repetido de plasma o concentrados comerciales purificados. Al no disponerse de antemano de una confirmación de laboratorio, en muchos casos se aplican estas transfusiones durante todo el período de duración de los episodios de trombosis o embolismo, asumiendo la sospecha diagnóstica por los antecedentes trombóticos personales o familiares. Idealmente, es aconsejable obtener las muestras sanguíneas para los análisis apropiados antes de la administración de transfusiones o de la terapia con heparina. La presencia de esta última en la circulación sólo invalida las pruebas de laboratorio funcionales, sin afectar apreciablemente la determinación del antígeno ATIII. Conviene resaltar que en los defectos puramente cuantitativos (disminución del antígeno ATIII, defecto tipo I), la tendencia trombótica ya empieza a manifestarse con reducciones relativamente moderadas (25-30%) por debajo de los valores hemostáticos usuales.
Factor Leyden (Resistencia a la proteína C activada)
Sobre la superficie de células endoteliales vasculares, y con la indispensable presencia de trombomodulina (TM), la acción enzimática de la trombina se encauza hacia la vía anticoagulante, mediante la activación de la proteína C. Las actividades enzimáticas procoagulantes clásicas de la trombina, independientes de la TM, consisten en la partición del fibrinógeno en fibrina y en intensificar la activación de los factores V, VIII y XIII. Esta proteína C activada (PCa), integrante principal de uno de los sistemas anticoagulantes fisiológicos naturales, ejerce su efecto neutralizante sobre los factores de coagulación V y VIII previamente activados (Va y VIIIa).
 A
partir de 1993 comenzaron a reportarse varias series de pacientes con episodios
de Trombosis Venosa Profunda que exhibían "in vitro" una resistencia
inusual del factor Va a la inactivación por la PCa. Con la ampliación
de estas investigaciones, se comprobó que este defecto es muy común
y que se expresa con gran frecuencia (hasta 15%) en varios países europeos.
La anormalidad genética responsable de este fenómeno se ubica
predominantemente (>90%) en una mutación puntiforme del factor V (FV
506Q), denominada factor "Leyden", por la ciudad en la cual fue inicialmente
descrita. Al menos 2 mutaciones adicionales recientemente descubiertas en otras
regiones de la molécula ("Cambridge" y "Hong Kong"),
también pudieran resultar trombofílicas, aunque por los momentos
su significación clínica es incierta (2).
A
partir de 1993 comenzaron a reportarse varias series de pacientes con episodios
de Trombosis Venosa Profunda que exhibían "in vitro" una resistencia
inusual del factor Va a la inactivación por la PCa. Con la ampliación
de estas investigaciones, se comprobó que este defecto es muy común
y que se expresa con gran frecuencia (hasta 15%) en varios países europeos.
La anormalidad genética responsable de este fenómeno se ubica
predominantemente (>90%) en una mutación puntiforme del factor V (FV
506Q), denominada factor "Leyden", por la ciudad en la cual fue inicialmente
descrita. Al menos 2 mutaciones adicionales recientemente descubiertas en otras
regiones de la molécula ("Cambridge" y "Hong Kong"),
también pudieran resultar trombofílicas, aunque por los momentos
su significación clínica es incierta (2).
En cuanto a la proclividad trombótica, el factor Leyden aparenta ser
la alteración más comúnmente asociada a Trombofilia. La
variedad heterozigota del alelo mutante induce un incremento de 5-10 veces del
chance de desarrollar trombosis venosa, mientras que en los que nacen homozigotos,
esta probabilidad aumenta unas 50-100 veces. Un estudio multicéntrico
sobre coincidencia entre trombosis venosa y factor Leyden detectó la
anomalía en >40% de pacientes incluidos por poseer antecedentes trombóticos
familiares y en un 20% de los casos no seleccionados, mientras que la incidencia
en la población normal correspondiente solo fue de 5% (2). La anormalidad
no conlleva a un riesgo aumentado de trombosis arteriales, a menos que se encuentre
asociada a hábitos tabáquicos u otros factores predisponentes,
en cuyo caso, estas coincidencias pueden facilitar la aparición de infartos
cardíacos en gente joven (7).
En proporción relativamente
alta el defecto Leyden coexiste con otras anomalías genéticas
de la coagulación, tales como las deficiencias de proteína C,
S, antitrombina y la hiperhomocistinemia. Cuando se presentan estas combinaciones,
el riesgo trombofílico puede magnificarse significativamente y generar
consecuencias clínicas severas. De ahí la importancia de ampliar
el espectro de las pesquisas de laboratorio al mayor número posible de
factores conocidos, cuando los indicadores clínicos así lo sugieran
(Tabla 2) (31).
Técnicamente, la constatación de resistencia a la PCa se ha simplificado
mediante la introducción de modificaciones en algunos procedimientos
básicos de coagulación, tales como el Tiempo Parcial de Tromboplastina.
Ocasionalmente, cuando las interpretaciones de los resultados proporcionados
por estas pruebas "simples" de despistaje son inconclusas, se recurre
a la demostración de la mutación específica (8). Esta contribución
de los análisis de biología molecular pueden requerirse también
para demostrar ciertos fenotipos adquiridos de resistencia a la PCa, no asociados
al factor Leyden. Tales situaciones han sido detectadas esporádicamente
durante el empleo de anticonceptivos orales, en presencia de Lupus Eritematoso
Diseminado o de un Anticoagulante lúpico asintomático y en asociación
con elevaciones marcadas del factor VIII.
Raramente se ha descrito
también una variedad de resistencia a la PCa de carácter familiar,
ajena al factor Leyden, lo que permite asumir la existencia de múltiples
variantes genéticas del propio factor V o de otros factores, que comparten
una misma expresión funcional (2).
La terapia anticoagulante coumarínica rutinaria no está indicada
en los pacientes heterozigotos, a menos que experimenten episodios trombóticos,
o quizás cuando exista una historia familiar extensa de tendencia trombótica.
Una vez diagnosticados, incluso preventivamente, los casos homozigotos deben
ser anticoagulados de por vida.
Factor
II (protrombina) G20210A
 Este
defecto representa una sustitución G A en el nucleótido
20210 del gen de la Protrombina. Su incidencia es solo ligeramente menor a la
del factor Leyden, lo cual, según la información más reciente,
la ubica como la segunda causa más frecuente de trombofilia hereditaria
(2), aun cuando su presentación homozigota es muy rara. La heterozigosidad
del alelo ha sido documentada en un 18% de pacientes cuyos familiares adolecen
de trombofilia, (solo 1% en grupos controles), y en un 6.2% de pacientes no
seleccionados que experimentaron un primer episodio de TVP, (2.2% de prevalencia
en controles). De manera similar a lo observado con el factor Leyden, su distribución
muestra predilección europea, más confluente hacia zonas mediterráneas.
Su ocurrencia no predispone regularmente a trombosis arteriales, aunque similarmente,
al coexistir con otras alteraciones puede acentuarse el riesgo de enfermedad
coronaria en gente joven. Casi todos los casos reportados cursan con una elevación
simultánea de la concentración de protrombina total, generalmente
en un rango de 20-30% por encima de los valores usuales.
Este
defecto representa una sustitución G A en el nucleótido
20210 del gen de la Protrombina. Su incidencia es solo ligeramente menor a la
del factor Leyden, lo cual, según la información más reciente,
la ubica como la segunda causa más frecuente de trombofilia hereditaria
(2), aun cuando su presentación homozigota es muy rara. La heterozigosidad
del alelo ha sido documentada en un 18% de pacientes cuyos familiares adolecen
de trombofilia, (solo 1% en grupos controles), y en un 6.2% de pacientes no
seleccionados que experimentaron un primer episodio de TVP, (2.2% de prevalencia
en controles). De manera similar a lo observado con el factor Leyden, su distribución
muestra predilección europea, más confluente hacia zonas mediterráneas.
Su ocurrencia no predispone regularmente a trombosis arteriales, aunque similarmente,
al coexistir con otras alteraciones puede acentuarse el riesgo de enfermedad
coronaria en gente joven. Casi todos los casos reportados cursan con una elevación
simultánea de la concentración de protrombina total, generalmente
en un rango de 20-30% por encima de los valores usuales.
Al ser reconocida la hiperprotrombinemia
como un factor independiente de riesgo trombótico arterial, se tiende
a imputar la acción trombogénica venosa, más bien a estas
elevaciones, que a la presencia de la molécula alterada. La demostración
inequívoca de esta alteración sólo se obtiene por las técnicas
genéticas clásicas de amplificación (PCR) y digestión
enzimática del ARN y la comparación con las sondas moleculares
apropiadas.
La experiencia general con esta anomalía todavía es limitada y
persisten algunas incógnitas en cuanto a su real significado clínico
y a las medidas terapéuticas aplicables a heterozigotos y homozigotos.
Es lógico suponer que durante episodios trombóticos, en casos
previamente diagnosticados, las transfusiones de plasma estarían justificadas,
aún cuando se desconoce el volumen, la frecuencia y el tiempo de administración
necesarios, debido a la imposibilidad de cuantificar el defecto secuencialmente.
Desde el punto de vista preventivo, no existe evidencia suficiente para recomendar
un régimen anticoagulante continuo.
Deficiencias de proteínas
C y S
Dentro del balance hemostático, estos componentes vitamina K dependientes, integran un sistema anticoagulante fisiológico de gran importancia, que limita los efectos procoagulantes de los factores activados V y VIII (Va, VIII) y dentro del cual, la proteína C es el actor glicoprotéico principal y la proteína S su cofactor enzimático.
La proteína C (PC) está conformada por una cadena liviana y una cadena pesada, que agrupan en conjunto un total de 417 aminoácidos. La cadena liviana contiene los dominios involucrados en la unión con fosfolípidos y calcio, así como 2 dominios homólogos al factor de crecimiento epidérmico (FCE). Dentro de la cadena pesada está incluida la acción de serino-proteasa y cuando es activada por la trombina, en presencia de TM, pierde un péptido, generándose de esta manera su capacidad inhibidora sobre los factores Va y VIIIa (7).
 La
concentración plasmática normal de proteína C es alrededor
de 3-5 mg/l y su deficiencia es demostrable en un 2-5% de pacientes con enfermedad
tromboembólica, aunque en personas mayores de 40 años con trombosis
recurrentes, la incidencia sube a 10-15%. Se han descrito cerca de 200 mutaciones
del gen de la PC asociadas con ausencias parciales o totales (defecto tipo I)
o alteraciones funcionales de la molécula (defecto tipo II). La primera
variedad es más frecuente, aunque no se han reportado diferencias en
la expresión clínica de las distintas mutaciones. Las presentaciones
habituales en los individuos heterozigotos incluyen la TVP, la Tromboflebitis
superficial y las complicaciones tromboembólicas pulmonares (9).
La
concentración plasmática normal de proteína C es alrededor
de 3-5 mg/l y su deficiencia es demostrable en un 2-5% de pacientes con enfermedad
tromboembólica, aunque en personas mayores de 40 años con trombosis
recurrentes, la incidencia sube a 10-15%. Se han descrito cerca de 200 mutaciones
del gen de la PC asociadas con ausencias parciales o totales (defecto tipo I)
o alteraciones funcionales de la molécula (defecto tipo II). La primera
variedad es más frecuente, aunque no se han reportado diferencias en
la expresión clínica de las distintas mutaciones. Las presentaciones
habituales en los individuos heterozigotos incluyen la TVP, la Tromboflebitis
superficial y las complicaciones tromboembólicas pulmonares (9).
Globalmente, el defecto induce un riesgo 7 veces mayor de desarrollar trombosis venosa y no incrementa significativamente la predisposición a sufrir de trombosis arteriales. La aparición de episodios veno-oclusivos tiende a aumentar con la edad y con el uso de contraceptivos orales. Curiosamente, aun cuando el embarazo estimula la síntesis de PC, en aquellas mujeres que padecen la deficiencia, las trombosis siguen siendo comunes, tanto durante el progreso del embarazo como en el período postparto.
La variedad homozigota de deficiencia de PC es rara, pero muy grave, e incide en la población general aproximadamente en 1 de cada 200.000-400.000 individuos. Se detecta casi sin excepción en el recién nacido, desde las primeras horas de vida, con un cuadro severo de "Púrpura Fulminante Neonatal", caracterizado por lesiones necróticas dérmicas reflejadas por un sustrato anatómico de oclusiones capilares. Se han documentado, además, trombosis cerebrales y oculares perinatales, que pueden iniciarse "in utero" durante las últimas semanas de la vida fetal. Ante la ausencia completa del sistema protector anticoagulante en el balance hemostático, la cascada de coagulación desinhibida suele condicionar, también, la aparición simultánea de fenómenos de Coagulación Intravascular Diseminada y lesiones multiorgánicas graves.
Este complejo sintomático sistémico resulta casi siempre fatal a corto plazo y la única posibilidad de tratamiento consiste en la administración de suficiente plasma fresco o de, según su disponibilidad, los concentrados comerciales de PC. Aún bajo terapia óptima la posibilidad de recuperación es mínima y una vez que el efecto transfusional desaparece, al persistir el déficit original, los episodios trombóticos tienden a reincidir con la misma intensidad.
Se ha comprobado que la disminución de la PC también facilita la aparición de los procesos de trombosis microvascular y Coagulación Intravascular Diseminada, las cuales acompañan a diversos cuadros sépticos y neoplásicos, notándose una estrecha relación entre la magnitud de la deficiencia y la gravedad de las manifestaciones clínicas respectivas (9). Como complicación adicional "sui generis" en muchos de estos casos, a las pocas horas de iniciada la terapia con Warfarina, aparece un fenómeno paradójico de severas lesiones necróticas de piel. Esta patología se origina por un imbalance transitorio, de breve duración, entre los mecanismos pro y anticoagulantes, debido a la relativa corta vida media de la PC (8 horas), en comparación con la más prolongada (20-24 horas), correspondiente a los factores procoagulantes cuya síntesis aun no ha sido inhibida suficientemente (factores II, IX, X).
 Durante
este breve período de imbalance crítico de inducción warfarínica,
a la preexistente reducción de la actividad funcional de la PC, se sobreponen
niveles aún inalterados de los otros factores; esta combinación
adversa incrementa la actividad procoagulante y permite la aparición
de trombosis microvasculares dérmicas.
Durante
este breve período de imbalance crítico de inducción warfarínica,
a la preexistente reducción de la actividad funcional de la PC, se sobreponen
niveles aún inalterados de los otros factores; esta combinación
adversa incrementa la actividad procoagulante y permite la aparición
de trombosis microvasculares dérmicas.
Algunos investigadores
pregonan la existencia de dos fenotipos bien diferenciables de la deficiencia
hereditaria de PC. En el primero, clínicamente dominante, los heterozigotos
son sintomáticos y hasta un 50% de sus familiares exhiben trombosis antes
de los 40 años; la prevalencia general se estima en 1/16.000 y se asume
que la forma homozigota es sumamente rara (9). En el segundo tipo, de herencia
recesiva, los heterozigotos son usualmente asintomáticos, con una incidencia
de 0.1 - 0.3% en donantes de sangre y solamente los homozigotos, o los heterozigotos
dobles, desarrollan complicaciones clínicas. Es interesante notar que
en distintos grupos estudiados, ambas variedades, dominantes y recesivos, exhiben
los mismos defectos genéticos (11).
La proteína S (PS) representa una glicoproteína relativamente
simple, integrada por 635 aminoácidos. Alberga dominios específicos
para su unión con el calcio y algunos fosfolípidos y una región
sensible a la acción de la trombina, relacionada estructuralmente con
el FCE. En el interior de esta última se realiza la acción enzimática
como cofactor de la proteína C. Presenta además una zona homóloga
a la globulina captante de hormonas sexuales, la cual se une a una fracción
proteica del complemento (proteína unida al C4b) (11).
Se atribuyen múltiples funciones a la proteína S, algunas aparentemente
extrahemostáticas:
Sólo la forma libre de la PS es capaz de actuar enzimáticamente como cofactor anticoagulante, mientras que la fracción combinada a proteínas es inerte. La concentración plasmática de la PS varía generalmente entre 20-25 mg/l, aunque tiende a ser muy fluctuante, incluso en condiciones fisiológicas.
En la población general, la deficiencia de PS aparenta ser menos común que la de PC, pero en pacientes con trombosis venosa las cifras de ambas tienden a equipararse. La herencia es autosómica dominante y los heterozigotos presentan una mayor predisposición a trombosis sintomáticas, calculándose que adolecen de un riesgo del 50% de enfermarse antes de los 45 años de edad (14). Los homozigotos cursan con cuadros clínicos muy severos, incluyendo la Púrpura Fulminante Neonatal, previamente descrita. La presentación de la deficiencia de PS exhibe características muy similares a la correspondiente deficiencia de PC, con la excepción de que un 5-13% de individuos heterozigotos pueden desarrollar, además, eventos trombóticos arteriales.
Existe una probable influencia de las hormonas sexuales femeninas sobre la síntesis de PS, ya que los niveles plasmáticos se encuentran disminuidos en mujeres menores de 45 años, en embarazadas y durante la ingestión de anticonceptivos orales.
Aparte de los defectos tipo I y II, una variedad especial (tipo III), quizás la más común, se caracteriza por exhibir una relación muy anormal entre sus fracciones circulantes, encontrándose la libre muy reducida y la ligada a proteínas relativamente elevada, sin alteraciones importantes en la concentración total. El tipo I y II parecen coexistir entremezclados en la gran mayoría de las familias estudiadas, por lo cual se los considera variedades fenotípicas de una misma patología genética. Todavía no se ha identificado alguna mutación específica asociada al tipo III.
Dada la diversidad de combinaciones fenotípicas posibles, el diagnóstico de laboratorio de ambas deficiencias debe incluir su determinación antigénica y funcional. El enfoque terapéutico está basado en la sustitución transfusional con plasma o concentrados comerciales durante las fases de trombosis aguda y la administración prolongada de Warfarina para los heterozigotos sintomáticos y los casos raros de homozigotos que logran sobrevivir.
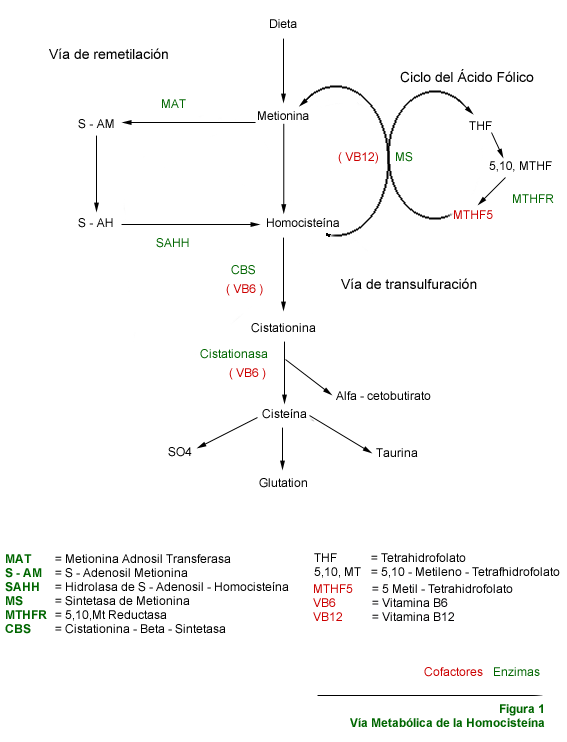
Hiperhomocistinemia (Homocistinuria)
La homocisteína se
genera como un metabolito intermediario del procesamiento de la metionina (aminoácido
esencial). Las reacciones bioquímicas y metabólicas pertinentes
a la homocisteína se encuentran esquematizadas en la Figura 1. Fundamentalmente
su acumulación excesiva ocurre por defectos en los mecanismos enzimáticos
responsables de su conversión final a cisteína y glutation. Durante
los procesos de metilación que van transformando la metionina ingerida
en homocisteína y cisteína, intervienen como cofactores importantes
los folatos, la piridoxina y la vitamina B12 (figura 1).
La asociación de la Homocistinuria Hereditaria severa con la aterotrombosis
prematura y las trombosis venosas profundas está bien documentada (15).
Se presume que la toxicidad trombogénica de la homocisteína pudiera
ejercerse a través de variados mecanismos que interfieren simultáneamente
sobre procesos fisiológicos de células endoteliales, plaquetas
y factores de coagulación (tabla 4). La deficiencia congénita
homozigota de la cistationina beta-sintetasa (CBS), de expresión autosómico-recesiva,
origina la patología más grave, típicamente diagnosticada
en la temprana infancia, y manifestada clásicamente por dislocación
del cristalino, glaucoma, retardo mental, osteoporosis, lesiones arteriales
ateroscleróticas e infartos múltiples, los cuales se evidencian
desde edades jóvenes. Otras deficiencias enzimáticas transmitidas
por herencia homozigota, tales como la de 5,10-MTHF, inducen hiperhomocistinemia
cuya severidad y consecuencias trombóticas son similares (34). La variante
heterozigota de la deficiencia enzimática de CBS cursa con elevaciones
menores de la homocisteína circulante y los cuadros clínicos respectivos
son más solapados y básicamente limitados al lecho vascular.
Se han identificado además defectos genéticos leves y de menor intensidad sintomática de algunas enzimas participantes en los pasos metabólicos esquematizados en la Figura 1. La homocistinemia ocasionada por estos defectos puede reducirse exitosamente con dosis altas de una combinación de las co-enzimas vitamínicas respectivas (tabla 4).
Por lo menos un 50% de los episodios trombóticos en la Homocistinuria Hereditaria ocurren en la circulación venosa (16), siendo la incidencia mucho menor en la deficiencia homozigota de MTHFR causada por la mutación c677 de su gen estructural (34). Las asociaciones con otras causas de trombofilia, especialmente con el factor Leyden, predisponen a una patología trombótica severa y recurrente. Las causas adquiridas de hiperhomocistinemia se describen en detalle en el párrafo correspondiente.
|
Endotelio
vascular
|
Plaquetas:
|
Coagulación
|
|
Generación
de agua oxigenada (cobre-dependiente) de toxicidad celular directa. |
Interferencia
en el metabolismo del ácido araquidónico con aumento de
la producción |
Aumento
de la síntesis del factor tisular |
|
Tabla
4 |
El fibrinógeno es una glicoproteína soluble que al gelificarse por efecto de la partición enzimática de la trombina, constituye el substrato del coágulo. Está integrado por 3 polipéptidos que incorporan un total de 1500 aminoácidos y codificado por la acción coordinada de 3 genes coparticipativos. Las alteraciones genéticas estructurales de la molécula pueden ocurrir en cualquier región de las secuencias lineares u originarse por alteraciones del ARN genómico o por defectos durante el proceso de transcripción. Ciertas modificaciones post-ribosomales, mucho menos frecuentes, pueden inducir además alteraciones de las zonas ocupadas por los carbohidratos.
 Hasta
la fecha de preparación de esta revisión, se han descrito mas
de 250 fibrinógenos cualitativamente anormales (disfibrinogenemias),
en los cuales la mayoría se origina por alteraciones en la secuencia
de aminoácidos, mientras que un grupo relativamente reducido exhibe deleciones
o inserciones segmentarias. Casi sin excepción, su presentación
genotípica es heterozigota y fenotípicamente predominan ampliamente
las variantes asintomáticas. Usualmente se descubren de manera accidental,
al investigar el significado de un tiempo de trombina marcadamente prolongado,
cuyo grado de alteración no guarda relación alguna con la incidencia
ocasional de manifestaciones hemorrágicas.
Hasta
la fecha de preparación de esta revisión, se han descrito mas
de 250 fibrinógenos cualitativamente anormales (disfibrinogenemias),
en los cuales la mayoría se origina por alteraciones en la secuencia
de aminoácidos, mientras que un grupo relativamente reducido exhibe deleciones
o inserciones segmentarias. Casi sin excepción, su presentación
genotípica es heterozigota y fenotípicamente predominan ampliamente
las variantes asintomáticas. Usualmente se descubren de manera accidental,
al investigar el significado de un tiempo de trombina marcadamente prolongado,
cuyo grado de alteración no guarda relación alguna con la incidencia
ocasional de manifestaciones hemorrágicas.
Existen situaciones excepcionales
en las cuales los fibrinógenos anormales se asocian con sangramientos
clínicos importantes, con deshicencia de heridas y, en mucho menor grado,
con una predisposición trombogénica. En este último grupo,
mayormente integrado por casos aislados se destacan los siguientes: "Argenteuil",
"Bergamo II","Chapel Hill I, III y V", "Copenhague",
"Dusard o Paris V", "Haifa", "Malmö", "Marburg",
"Milano II o Naples", "New Albany", "New Orleans II,
"New York I", "Nijmegen", "Oslo I", "Poitiers"
y "Richfield". Por convención internacional esta nomenclatura
se establece según el sitio de identificación, o de acuerdo al
apellido del individuo o grupo familiar afectado.
Como es de suponer, estas mutaciones conforman un conglomerado interesante de
errores genéticos, que han contribuido a dilucidar las interacciones
fisiológicas trombina-fibrinógeno. Pero, dada su rareza, al encontrarse
limitadas a una sola persona, o a pocos miembros de una misma familia, poseen
poca repercusión clínica en el despistaje rutinario de trombosis.
Para explicar la tendencia trombótica de estos fibrinógenos anormales,
algunos experimentos in vitro sugieren las siguientes posibilidades:
El diagnóstico se sospecha inicialmente con el descubrimiento de un tiempo de trombina o protrombina alargados, mientras que la confirmación de la disfunción hemostática se obtiene por medio de métodos que evalúan la capacidad y velocidad de polimerización de las cadenas polipeptídicas. Una vez demostradas las anormalidades de polimerización, en los centros especializados en biología molecular, se prosiguen los estudios para demostrar el tipo de mutación y su ubicación exacta en alguna de las 3 cadenas polipeptídicas. Finalmente, para cooperar en la catalogación internacional se cotejan las particularidades moleculares encontradas con las correspondientes a las disfibrinogenemias previamente descritas.
Alteraciones del sistema fibrinolítico
La función esencial de la fibrinólisis fisiológica reside en la disolución de los coágulos de fibrina que continuamente se van acumulando en la microcirculación, debido a la activación, normalmente muy restringida, de la cascada de coagulación. La plasmina, originada por el fraccionamiento enzimático del plasminógeno por activadores endoteliales, es la enzima responsable en disolver los coágulos, aunque su acción proteolítica también abarca las moléculas de fibrinógeno y algunos factores de la coagulación. Es lógico asumir que dentro del balance hemostático natural una actividad fibrinolítica deficiente, congénita o adquirida pudiera favorecer la aparición de trombosis o, al menos, prolongar su curso evolutivo.
Los defectos del sistema fibrinolítico pueden originarse por:
 La
repercusión clínica de estas alteraciones, sobre todo de aquellas
relativas al plasminógeno ha sido muy debatida. Varios grupos de investigadores
han reportado hipoplasminogenemias individuales o familiares, de transmisión
heterozigota, con características claras de herencia autosómica
dominante, las cuales aparecen indudablemente asociadas con una alta proporción
de trombosis venosas espontáneas o post-quirúrgicas (25,26). Otros
autores interpretan opuestamente hallazgos similares de laboratorio, negando
por completo su importancia trombogénica (27). Dentro del panorama general
de las condiciones trombofílicas, la incidencia de estos defectos parece
insignificante. En las escasas series analizadas, las manifestaciones trombóticas
no son disímiles de las observadas en otras causas de trombofilia, aunque
se ha sugerido que algunas localizaciones muy atípicas, tales como las
trombosis de venas retinianas y de senos venosos cerebrales, o los abortos repetidos
por trombosis placentarias, pudieran señalar la presencia de este tipo
de defecto (25). La confirmación de laboratorio no es muy complicada
y, en particular, la dosificación del plasminógeno y las pruebas
generales de fibrinólisis son de fácil ejecución.
La
repercusión clínica de estas alteraciones, sobre todo de aquellas
relativas al plasminógeno ha sido muy debatida. Varios grupos de investigadores
han reportado hipoplasminogenemias individuales o familiares, de transmisión
heterozigota, con características claras de herencia autosómica
dominante, las cuales aparecen indudablemente asociadas con una alta proporción
de trombosis venosas espontáneas o post-quirúrgicas (25,26). Otros
autores interpretan opuestamente hallazgos similares de laboratorio, negando
por completo su importancia trombogénica (27). Dentro del panorama general
de las condiciones trombofílicas, la incidencia de estos defectos parece
insignificante. En las escasas series analizadas, las manifestaciones trombóticas
no son disímiles de las observadas en otras causas de trombofilia, aunque
se ha sugerido que algunas localizaciones muy atípicas, tales como las
trombosis de venas retinianas y de senos venosos cerebrales, o los abortos repetidos
por trombosis placentarias, pudieran señalar la presencia de este tipo
de defecto (25). La confirmación de laboratorio no es muy complicada
y, en particular, la dosificación del plasminógeno y las pruebas
generales de fibrinólisis son de fácil ejecución.
Una vez establecida, la trombosis se trata de manera convencional. La posibilidad de usar fibrinolíticos parenterales en casos definitivamente comprobados de deficiencia o alteraciones del plasminógeno aun no han sido evaluadas.
Aumento del factor VIII (factor antihemofílico A)
Este conocido factor, cuya deficiencia origina el síndrome clásico de Hemofilia "A", ha sido incorporado recientemente a la lista de trombofilia. Varios estudios prospectivos han comprobado que su aumento constituye un factor de alto riesgo independientemente de otros concomitantes. En una de las casuísticas más recientes (32) en 360 pacientes consecutivos que habían padecido un solo episodio de TVP, se observó que a niveles del factor VIII encima del percentil 90 de lo normal, el riesgo se incrementa 7 veces.
En contraste con los resultados obtenidos para el factor Leyden, la relación factor VIII/trombosis no es directamente linear y la posibilidad de recurrencia trombótica varía entre 5-20% por año (33). Parece muy probable que cuando las concentraciones superan las 150-175 u/dl la incidencia de trombosis empieza a elevarse apreciablemente. Se desconocen los estímulos que determinan un aumento de la síntesis del factor VIII, pues no existe una relación aparente con las reacciones de fase aguda.
Teóricamente se invoca una predeterminación genética, aunque desconocemos las oscilaciones naturales de los niveles plasmáticos durante el ciclo vital, empezando por la infancia, e ignoramos lo que ocurre secuencialmente en los meses o años después de uno o varios episodios de trombosis. (32). Una vez transcurrido el período usual de terapia anticoagulante post-trombótica (habitualmente 3-6 meses), en ausencia de recurrencias, y en vista de que aún no se han efectuado los estudios prospectivos adecuados, tampoco existe suficiente evidencia para justificar su empleo profiláctico más prolongado o indefinido.
![]()