

Bases Genéticas del SQTL
El síndrome de QT largo congénito es causado por la mutación de al menos 5 locus genéticos de canales iónicos ubicados en los cromosomas 3,4,7,11 y 21 (7-8) . La mutación del gen del canal de sodio y 3 genes de canales de potasio han sido identificados , siendo reportados hasta ahora más de 120 mutaciones (9) :
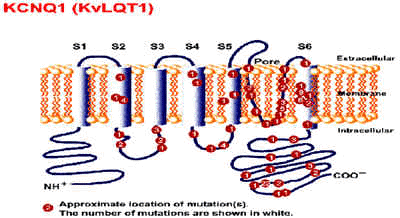
Mutación del gene KVLQT1 ,en el cromosoma 11p15.5 (LQT1) , si es homocigótica produce el síndrome de Langen Nielsen, si la mutación es heterocigótica produce el síndrome de Romano Ward . KVLQT1 codifica una proteína del canal de potasio Iks , que interactua con otro pequeño canal de potasio el Mink para formar el canal de potasio Iks . Las mutaciones de éstos, originan pérdida en la función del canal . Esta es la variante más frecuente.
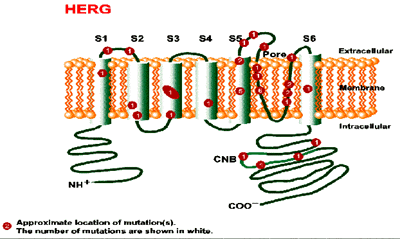
Mutación del gen HERG (LQT2) en el cromosoma 7q35-36 .. HERG (LQT2) codifica al canal Ikr de potasio , produciendo su mutación una pérdida de la función del canal . Un aumento de la concentración plasmática de potasio puede originar aumento en la salida de potasio y acortar efectivamente el QT en pacientes con esta mutación
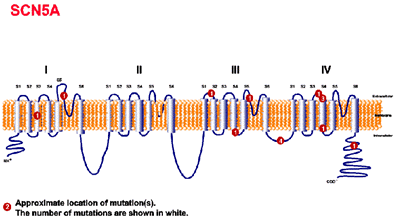
Mutación del gen SCN5A (LQT3) en el cromosoma 3p21-24 . El SCN5A codifica el canal cardíaco de sodio por lo cual, su mutación origina una fase tardía de inactivación con resistencia de la pared celular a corrientes de entrada . La mutación origina un aumento de la función del canal INa . El Mexiletine , un bloqueante de canales de sodio, es eficaz en acortar el QT en pacientes con esta alteración aún cuando su administración no excluye el uso de betabloqueantes
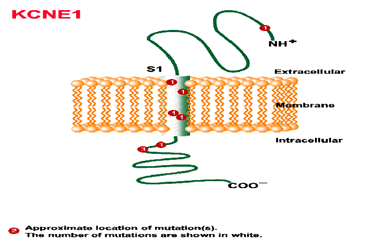
Mutación del gen MinK (LQT5) en el cromosoma 21q22 codifica una subunidad del canal Iks.
El gen del LQT4 no ha sido identificado.
. Existen tres variantes del SQTL congénito :
El síndrome de Langer-Nielsen se transmite de manera autosómica recesiva , pudiendo los familiares portar la alteración aún siendo asintomáticos.
El síndrome de Romano Ward es autosómico dominante
Zareba y col (10) demostraron que el genotipo del síndrome de QT largo influye en el curso clínico . El riesgo de eventos cardíacos (síncope , muerte súbita) es mayor en los pacientes con mutaciones LQT 1 o LQT2 comparado con el grupo de pacientes LQT3 , sin embargo, cuando se presenta alguna manifestación clínica en grupo LQT 3 el porcentaje de eventos fatales es mayor que en los otros grupos . La posibilidad de muerte durante un evento cardiovascular en el grupo LQT3 es de 20 % , siendo de 4 % en los otros grupos.
Priori y col (7)sugieren que pudiese existir diferente penetración de acuerdo al grado de mutación del KVLQT1 en la población general, lo cual pudiese explicar la sensibilidad a arritmias ventriculares inducidas por medicamentos en la población general. En otro estudio el mismo Priori (11) muestra que la penetración no siempre es alta (alrededor de 90%) como se planteaba normalmente , sino, que en algunas familias la penetración es baja (25%) . Estos pacientes pueden presentar una alta predisposición a arritmias ventriculares aún con QT normal , por lo cual, en familiares de pacientes con QT largo deberá realizarse una evaluación completa y no sólo una evaluación clínica para establecer su riesgo.
![]()