
![]()
Tumor
Fibroso Solitario | Tumores
del Estroma Gastrointestinal
| Leiomiosarcomas
| Fibrohistiocitomas benignos y de Malignidad intermedia
Fibrohistiocitoma Maligno y Sarcomas de Alto Grado
| Tumores del tejido adiposo
| Hemangiopericitoma y tumor glómico |
Tumores vasculares
Discusión
Tumor
Fibroso Solitario
Entre los tumores fusocelulares descritos en los resultados de esta investigación,
discutiremos inicialmente los problemas que se presenta para precisar el diagnóstico
del tumor fibroso solitario (TFS), una neoplasia de células fusiformes
descrita en el año 1931 como un tumor pleural (17) y posteriormente observado
en el mediastino, en el pericardio e intrapulmonar (18). Más recientemente
se demostró el TFS en localizaciones extratoráxicas y en este
trabajo precisamente presentamos varios ejemplos de TFS intrabdominal o en el
retroperitoneo (19,20). Una de las particularidades de esta neoplasia es la
cantidad de variaciones en su apariencia histológica; en el TFS alternan
áreas fusocelulares con fibrosis e hialinización, apariencia mixoide
y en ocasiones hemangiopericítica y presencia de áreas con discreto
pleomorfismo, mitosis usualmente típicas y necrosis focal por lo que
en estos casos es difícil predecir su evolución y pronóstico.
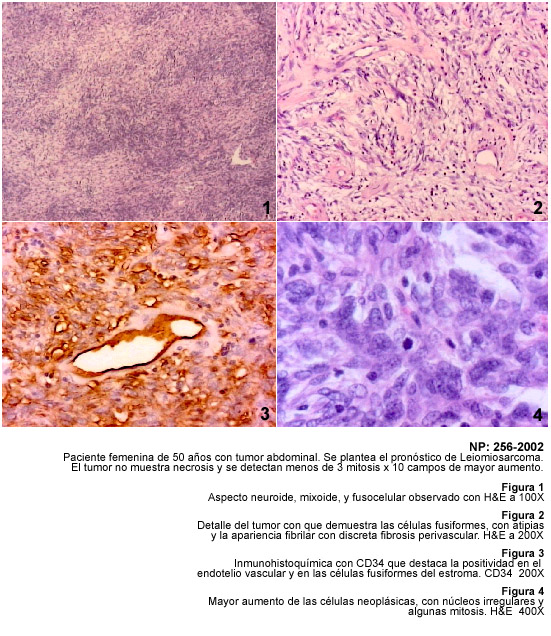
La demostración de TFS en sitios extratoráxicos y los cambios histológicos sugestivos de malignidad, como mitosis y necrosis, han sido descritos previamente y se señalan a propósito de las características patológicas de nuestros casos (21,22). La investigación inmunohistológica de los casos de TFS destaca las dificultades que existen para el diagnóstico macroscópico e histopatológico y la ayuda de la inmunohistoquímica con el anticuerpo CD34 para hacer un adecuado diagnóstico diferencial.
La variabilidad histológica del TFS obliga a plantear el diagnóstico diferencial entre diversos tumores mesenquimáticos como el leiomiosarcoma, fibrosarcoma, sarcoma sinovial monofásico, hemangiopericitoma, tumores malignos de la vaina de los nervios periféricos, schwannoma y los tumores del estroma gastrointestinal. Por otra parte el TFS muestra áreas con aspecto de hemangiopericítico y puede simular un timoma fusocelular, un carcinoma fusocelular, un dermatofibrosarcoma protuberans e incluso un mesotelioma desmoplásico. La aplicación de la técnica de inmunohistoquímica para diagnosticar el TFS no solo depende de CD34 sino del uso de Vimentina, y de diversos anticuerpos como Actina muscular, Proteina S-100, PGP 9.5, Queratinas, Desmina y otros. El TFS es positivo a la Vimentina, y a CD34, y se ha demostrado inmunomarcaje con bcl 2; algunos tumores pueden ser focalmente positivos a S-100 y también se ha descrito positividad para citoqueratina y para Desmina, posiblemente representando una diferenciación miofibroblástica en estos tumores (22). Los criterios de malignidad en el TFS siguen siendo la hipercelularidad, la necrosis, atipias nucleares y un recuento mitósico mayor de 10 mitosis por campo de mayor aumento (20,23).
Todos estos tumores demostrados, están señalados en la Tabla I con diagnóstico de TFS.
Tumores
del Estroma Gastrointestinal
En la cavidad abdominal, usualmente adheridos a las paredes del tubo digestivo,
se pueden ver neoplasias fusocelulares de origen diverso, pero entre ellas,
las más comunes son los tumores del estroma gastrointestinal, abreviados
con las iniciales GIST (24,25). Más de la mitad de los GIST se ven en
el estómago, de 20 a 30% en el intestino delgado y un 10% en el esófago,
el colon y el recto, usualmente aparecen después de la cuarta década,
sus células son fusiformes o pueden adoptar una apariencia epitelioide
con arreglos histológicos de una gran variabilidad como fasciculada,
en forma de estoril, con aspecto alveolar, en empalizada y hemangiopericitoide,
por lo que el diagnóstico diferencial debe hacerse con el TFS, fibrosarcoma,
leiomiosarcoma, schwannoma y tumor maligno de la vaina de los nervios periféricos.
En algunas ocasiones, sus células adoptan una configuración oncocítica,
rabdoide o granular lo cual aumenta las dificultades del diagnóstico.
La inmunohistoquímica de los tumores GIST revela que de 70 a 83% de ellos reaccionan positivamente con CD34, de 20 a 40% marcan con Actina muscular específica, 30 a 50% con Desmina, algunos son positivos a la Proteína S-100 y entre el 86 y el 100% inmunoreaccionan con c-kit (CD117) un anticuerpo que identifica las células intersticiales de Cajal (26, 27).
Los GIST han sido subclasificados en tumores de los nervios autonómicos
gastrointestinales (GANT), en ocasiones difícilmente diferenciables de
los tumores GIST ya que a pesar de ser algo más vascularizados, la morfología
y la inmunohistoquímica es similar por lo que algunos han sugerido el
uso del microscopio electrónico para precisar su diagnóstico (25).
Aproximadamente de un 25 a 30% de los GIST muestran caracteres de malignidad
y por ello, algunos prefieren hablar de Sarcomas del Estroma Gastrointestinal
(GISS); estos tumores recurren, y pueden dar metástasis a los pulmones,
al peritoneo y al hígado. Recientemente, se ha señalado que no
existen criterios precisos para predecir el comportamiento maligno de los tumores
GIST (28). Por esta razón, se ha propuesto el uso con inmunohistoquímica
con marcadores de proliferación nuclear como PCNA y Ki 67 para investigar
sobre el pronóstico (29). Sin embargo, es difícil asegurar sobre
como evolucionarán los GIST con caracteres de malignidad (24).
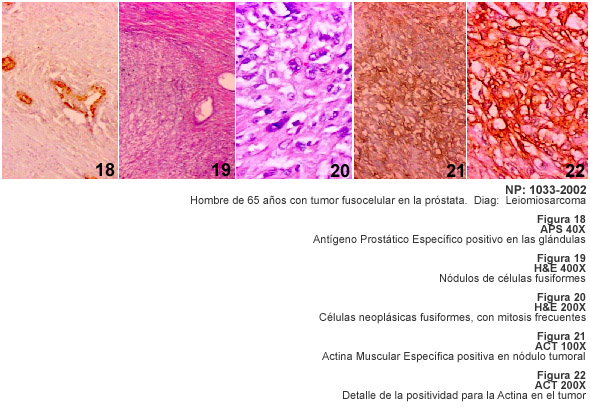
Leiomiosarcomas
Es importante recordar que los tumores de músculo liso con apariencia
epitelioide se ven no solamente en el tracto gastrointestinal, sino también
en otras localizaciones como el útero, retroperitoneo, vagina, subcutis,
etc., y en el caso de los leiomiomas, leiomiomas atípicos y leiomiosarcomas
uterinos es importante el uso de CD34 para diferenciarlos de los sarcomas del
estroma endometrial (30).
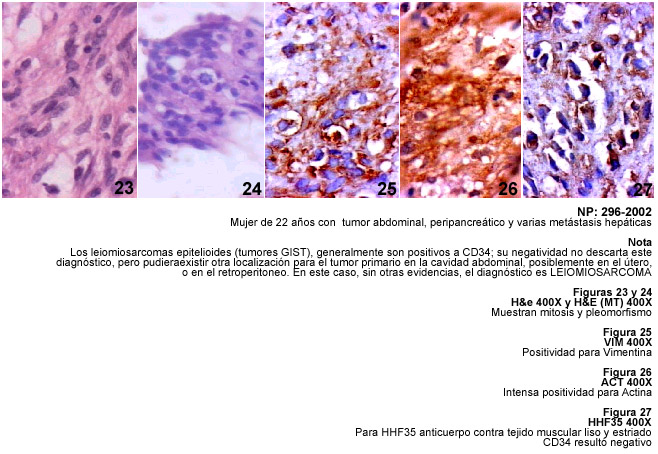
La expresión de CD34 y focalmente de queratina en un número significativo de miomas de músculo liso y de leiomiosarcomas, demuestran características parcialmente compartidas con los tumores GIST y sirven para diferenciarlos de tumores epiteliales, no así de los sarcomas del estroma endometrial, aunque estos rara vez son positivos a CD34. Recientemente se ha descrito en las neoplasias del estroma endometrial la presencia de CD10 (CALLA), un antígeno que caracteriza a las células de la leucemia linfoblástica (31).
Fibrohistiocitomas
benignos y de Malignidad intermedia
En este trabajo examinamos un Dermatofibroma, también llamado Fibrohistiocitoma
Benigno Cutáneo (FHBC). Estos tumores pueden tener diversos aspectos
histológicos, pueden ser difusos y más a menudo nodulares, usualmente
subcutáneos, muestran células fusiformes que alternan con pequeños
vasos, algo de hemosiderina y el tejido conectivo muestra áreas verticiladas.
También dentro de los FHBC pueden verse otras variantes como el hemangioma
esclerosante, el FHBC Epitelioide, el FHBC Atípico o pseudosarcomatoso,
algunos FHBC con aspecto de empalizada que simulan ser de origen neuroide en
los que la Proteína S-100 demuestra ser negativa y el Fibrohistiocitoma
Angiomatoide o aneurismático, del cual hemos incluido también
un caso en esta serie. La importancia de CD34 en estos tumores deriva de la
negatividad de este anticuerpo en las células tumorales, observándose
el inmunomarcaje característico solamente en las paredes vasculares.
El Fibrohistiocitoma Aneurismático o Angiomatoide (FHA), es un FHBC que
puede ofrecer algunos problemas ya que su presentación clínica,
es a menudo como un área pigmentada por lo que se confunde con un nevus.
En ellos pareciera existir un rápido crecimiento debido a las hemorragias
o a traumatismo sobre el tumor, donde se producen lagos de sangre por debajo
de la epidermis y por ello puede también ser interpretado como un melanoma.
Histológicamente, la apariencia de FHA muestra células fusiformes,
algunas atípicas, muchas multinucleadas, histiocitos con abundante hemosiderina
y un estroma celular, a menudo verticilado, que rodea espacios llenos de eritrocitos.
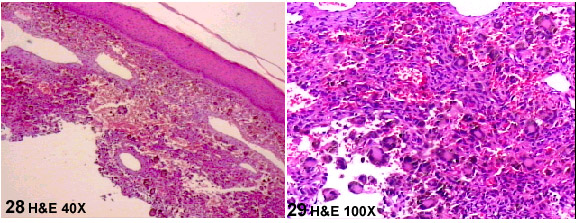
Este tumor mesenquimático benigno, fue descrito por Santa Cruz y Kyriakos
en 1981 (32) y en sus células no se observa positividad con CD34, pero
este inmunomarcaje sirve para demostrar su rica vascularización. Ver
las Figuras 28 y 29 de uno de los casos recibidos en Novapath: Hombre de 41
años que dice tener una "placa melánica" en la región
xifoidea, que le ha crecido súbitamente. Le hacen una biopsia superficial.
Se confunde con un Melanoma por la hemosiderina.
La apariencia del FHA es siempre típica, con lagos de sangre y no debe ser confundido el diagnóstico con FHM o con Melanoma. El caso NP:618-2001 de una mujer de 45 años con una lesión en la pierna izquierda es histológicamente idéntico al demostrado en la microfotografías anteriores.
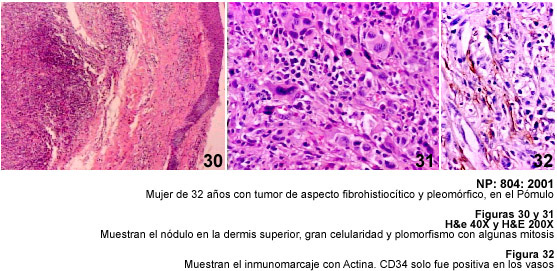
Entre los Fibrohistiocitomas
cutáneos de malignidad intermedia hemos incluido un caso de Fibroxantoma
Atípico (FXATP) y cinco casos de Dermatofibrosarcoma protuberans (DFSP).
El FXATP es una lesión de la piel afectada por el sol, y por lo tanto
se observa en la cara. Nuestro caso era una lesión en el pómulo,
y puede verse también en el cuello, el tronco y las áreas proximales
de las extremidades, usualmente en personas mayores o en sujetos que presentan
alguna cicatriz por quemadura o por efecto de radiación. Los tumores
infiltran la dermis superior y siendo circunscritos, en la periferia se observa
compresión de los anexos de la piel y distorsión por la proliferación
de células fusiformes, atípicas, que pueden ser pleomórficas
y con mitosis. Ante una lesión de la piel con estas características,
uno de los diagnósticos planteables es el carcinoma fusocelular, o un
melanoma. La inmunohistoquímica ayuda a descartar un tumor epitelial
con el uso de las queratinas en cocktail (AE1/AE3), y solo existe positividad
con la Vimentina en sus células, pues con los anticuerpos utilizados
no se demuestra inmunomarcaje con CD34, ni con la Proteína S-100 (33,
34).
El DFSP es un tumor que se puede observar en el tronco, cabeza y cuello y en el extremo proximal de las extremidades superiores e inferiores. En ocasiones, se ha relacionado con traumatismos previos o con áreas de cicatrices y mide entre 2 y 5 cms, su crecimiento es lento, pueden mostrar varios nódulos, uno solo, o placas en la dermis superior y algunas veces profunda. La característica más relevante del DFSP es su apariencia histológica repetitivamente verticilada, creando remolinos, especie de estoriles de células fusiformes con pequeños vasos centrales. El pleomorfismo y las atipias son raros, pero pueden verse mitosis, usualmente no más de 5 por 10 campos de mayor aumento (35). Existe una variedad pigmentada del DFSP, el denominado Tumor de Bednard en el cual se describen melanocitos entre las células fusiformes y estos pueden ser inmunomarcados con la Proteína S-100 y con HMB45 (36, 37). El inmunomarcaje positivo para CD34 es una de las características resaltantes del DFSP, por ello, este tumor de malignidad intermedia puede fácilmente diferenciarse de otros histiocitomas benignos o malignos (38). Debe siempre considerarse en el diagnóstico diferencial del DFSP a los tumores de estirpe nerviosa, especialmente por su apariencia fusocelular y fibrilar y por la posibilidad de positividad para la Proteína S-100. En esta situación, debe señalarse que los Tumores Malignos de la Vaina de los Nervios Periféricos, también positivos a la Vimentina y a S-100, son frecuentemente positivos a CD34 (7). La posibilidad de que el DFSP presente áreas de Fibrosarcoma es un hecho descrito (39). En estos casos, el pleomorfismo, la celularidad y la actividad mitótica aumentan y pueden producirse metástasis (40). En nuestros casos hemos incluido un DFSP de la piel de la mama, situación que hemos visto en varias mujeres en quienes se ha planteado el diagnóstico diferencial con tumores mamarios. Esta ubicación del DFSP también ha sido documentada previamente (40, 41, 42).
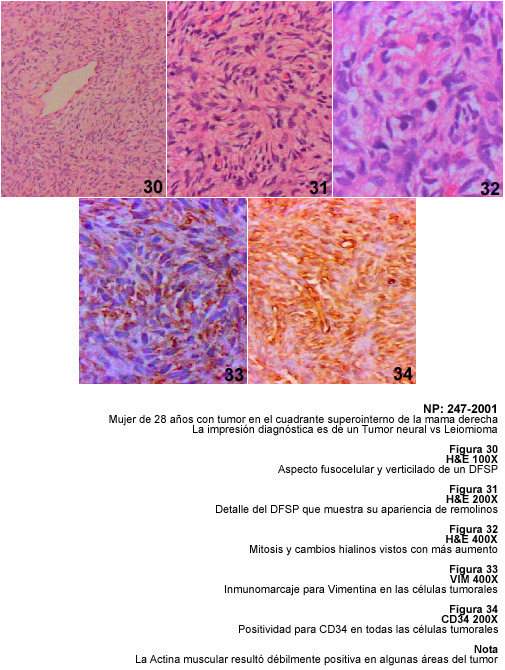
Fibrohistiocitoma
Maligno y Sarcomas de Alto Grado
La inclusión de los Fibrohistiocitomas Malignos (FHM) y de los Sarcomas
de Alto Grado en este grupo, de neoplasias examinadas inmunohistoquimicamente
con CD34 y con otros anticuerpos, se debe a la necesidad de revisar el diagnóstico
diferencial de los tumores fusocelulares, en particular cuando estos muestran
criterios de malignidad. En este sentido, ya hemos señalado como el TFS
puede presentar variantes malignas. Igual sucede con los tumores de músculo
liso y los tumores GIST, sobre todo si existe pleomorfismo. Más adelante
discutiremos el inmunomarcaje del Lipoma Pleomórfico y del Liposarcoma
Pleomórfico y ya hemos señalado como otras neoplasias de malignidad
intermedia como el DFSP pueden tener áreas sarcomatosas. Estudiamos seis
casos de FHM y cuatro casos con diagnóstico de Sarcoma de Alto Grado,
en dos de ellos con ciertas evidencias sobre su histogénesis, supuestamente
miógena y neuroide, sobre la base de la inmunohistoquímica, sin
que pudiesen esos tumores ser clasificados como Leiomiosarcoma o rabdomiosarcoma,
ni como sarcoma neurogénico, pero tampoco como FHM. Recientemente, se
demostró como muchos de estos Sarcoma de Alto Grado en los adultos son
Rabdomiosarcomas Pleomórficos (43) y ellos usualmente son clasificados
como FHM, LMS, Liposarcomas Pleomórficos o sencillamente Sarcomas de
Alto Grado (44,45).
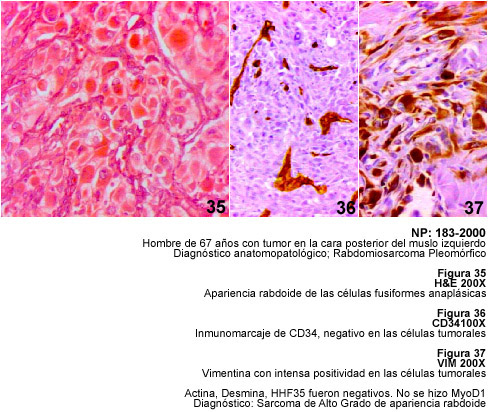
El concepto del llamado FHM fue propuesto por Stout a comienzos de los años 60 (46,47) y el mismo se consolidó con la publicación de numerosos casos describiendo esta neoplasia pleomórfica, la cual ha venido a constituir hoy día - con sus variantes, mixoide, de células gigantes, inflamatorio y hasta el llamado FHM angiomatoide que no encaja dentro del grupo - uno de los sarcomas más comunes de los adultos (48). El pleomorfismo, es uno de los conceptos que se usan para el diagnóstico de estas neoplasias puesto que no existen marcadores inmunohistoquímicos ni criterios ultraestructurales específicos para hacer el diagnóstico de FHM (3,49,50, 51). Fletcher ha demostrado como la revisión de 159 casos diagnosticados como FHM ha servido para reclasificar el 25% de ellos como liposarcomas (34 pleomórficos y 6 desdiferenciados) y el 18.2% como Leiomiosarcomas (52). Un hecho tranquilizador a la luz de los más recientes avances puede ser la irrelevancia del pleomorfismo en lo que se refiere al pronóstico y al tratamiento de los sarcomas de alto grado y del FHM, no así cuando el diagnóstico diferencial planteado es con un linfoma anaplásico, con un carcinoma indiferenciado o con un melanoma, tumores estos también caracterizados histológicamente por marcado pleomorfismo celular. En los casos antes señalados, la ayuda de la inmunohistoquímica para el diagnóstico y la decisión terapéutica es fundamental (5).

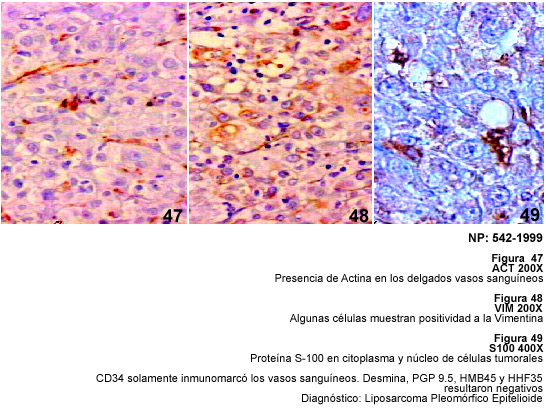
Tumores
del tejido adiposo
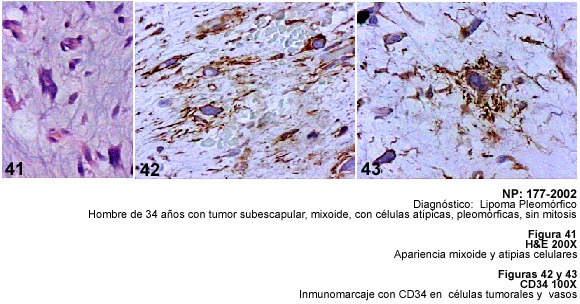
Cuando se examina el anticuerpo CD34 en los tumores del tejido adiposo, se puede
destacar el arreglo vascular de los Liposarcomas mixoides e identificar la presencia
de células fusiformes y dendríticas perivasculares descritas por
Suster y Fisher en los Liposarcomas (15). Deben diferenciarse estas células
de los adipocitos neoplásicos que son positivos a la Proteína
S-100, y que no reaccionan con CD34 a diferencia de los adipocitos atípicos
bien sea como floretas o como células multinucleadas atípicas
observadas en los Lipomas Pleomórficos y en los Lipomas Fusocelulares,
por lo que en estos casos se puede plantear el diagnóstico diferencial
con el TFS (53). También se ha propuesto que los Lipomas Fusocelulares
son neoplasias de células dendríticas intersticiales más
que verdaderos Lipomas (15). En general, los Liposarcomas pueden ser bien diferenciados
y desdiferenciados, mixoides y de células redondas, y los Liposarcomas
Pleomomórficos (LSP). Estos últimos, a pesar de ser neoplasias
poco frecuentes, su diagnóstico diferencial se debe hacer con los Sarcomas
de Alto Grado, para lo cual la inmunohistoquímica pude ser de gran ayuda
(54). El parecido del LSP con el Fibrohistiocitoma Maligno (FHM) ha sido discutido
ampliamente por Fletcher (52) quien en 159 tumores diagnosticados como FHM encontró
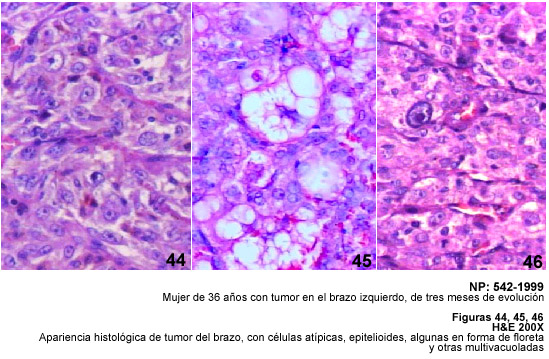
34 casos (21%) que se reclasificaron como LSP. El caso de LSP que se examina
en este trabajo, es el único que no pertenece a la casuística
de los años 2000 al 2002, pues fue recibido en Novapath a finales de
1999. La decisión de incluirlo en nuestra serie, se debe a la importancia
del mismo para discutir el tema del pleomorfismo celular en los sarcomas y del
uso de CD34 en el diagnóstico. El LSP se encontraba en el brazo y fue
considerado como una variante epitelioide del mismo, lo cual hacía más
complicado su diagnóstico (55). La positividad para S-100 con negatividad
para CD34 apoya el diagnóstico de LSP y de acuerdo con lo que hemos descrito
previamente, CD34 sirve para diferenciarlo del Lipoma Pleomórfico. En
la discusión de los tumores fibrohistiocíticos hemos hecho énfasis
en el pleomorfismo de los fibrohistiocitomas y de los sarcomas de alto grado,
y hemos comparado estos tumores con neoplasias pleomórficas del tejido
adiposo. Llamamos nuevamente la atención sobre la positividad para CD34
en el Lipoma Pleomórfico, un tumor que puede confundirse con el LSP,
e insistimos que al ser inmunomarcadas sus células atípicas con
este anticuerpo, se facilita el diagnóstico (15).
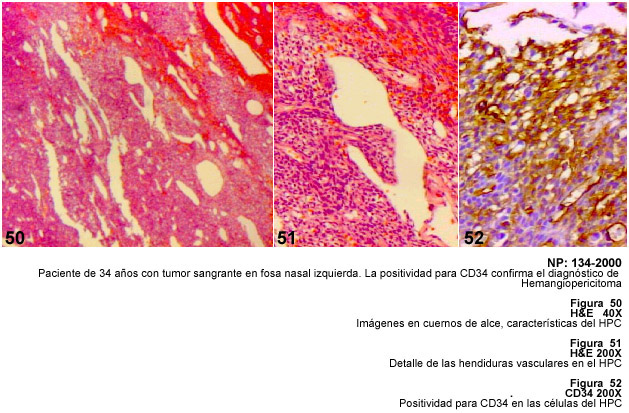
Hemangiopericitoma
y tumor glómico
Los hemangiopericitomas (HPC), que fueron descritos inicialmente por Stout y
Murray como tumores que se originan de los pericitos (56) y estudios ultraestructurales
(57) e inmunohistoquímicos (58,59), demostraron posteriormente este hecho.
Recientemente, se han descrito marcadores de proliferación celular para
investigar con inmunohistoquímica el pronóstico de estas neoplasias
(60). Las dificultades con el HPC se producen por la apariencia hemangiopericítica
que muestran muchos tumores fusocelulares como son el sarcoma sinovial, el TFS,
el FHM, los angiosarcomas y el condrosarcoma mesenquimático. Por otra
parte, existen áreas mixoides, alveolares y trabeculares en los HPC,
que pueden confundir el diagnóstico con rabdomiosarcoma y con liposarcoma
mixoide. La positividad para el anticuerpo CD34 y la Vimentina con negatividad
para CD31, citoqueratinas y para la Proteína S100, caracterizan al HPC,
sin descartar casos con débil positividad para la Actina Muscular específica.
En este trabajo presentamos dos tumores glómicos y tres HPC, uno de ellos
de las fosas nasales. Estas dos neoplasias, el tumor glómico de estirpe
muscular y los HPC originados en los pericitos, son considerados dentro de los
tumores benignos perivasculares en la reciente edicción del fascículo
de los Tumores de los Tejidos Blandos del AFIP (61), por lo que los hemos examinados
juntos en esta investigación. Los HPC de los senos paranasales son frecuentemente
benignos a diferencia de los HPC de las extremidades y del retroperitoneo que
son más agresivos, usualmente mayores de 5 centímetros, hipercelulares,
con más de 4 mitosis por campo de mayor aumento, con pleomorfismo, focos
de hemorragia y necrosis (62,63). La invasión vascular, las mitosis y
las células gigantes se relacionan con la agresividad del tumor (60).
Presentamos también en este estudio un HPC de las meninges destacando la negatividad de este tumor para CD34 y aprovechando esta particularidad para recordar que esta neoplasia fue considerada inicialmente como una variante del meningioma angioblástico (64) y que posteriormente se aceptaron los criterios de Stout y Murray (56) y se le consideró como HPC, similar al de los tejidos blandos. Recientemente, estudios comparativos hechos en Japón (65) señalan un origen diferente para los HPC de las meninges y los de los tejidos blandos, hallazgos estos que apoyarían la negatividad para CD34 señalada en nuestros resultados.
Tumores
vasculares
El aporte de la microscopía electrónica y de la inmunohistoquímica
al diagnóstico de los tumores vasculares, gira alrededor de los cuerpos
de Weibel-Palade, estructuras citoplasmáticas donde reside el factor
de Von Willebrand (Factor VIII) el cual puede observarse inmunohistoquímicamente
en el endotelio vascular (3, 5). CD31 es un anticuerpo usado para marcar el
endotelio vascular y es más específico que el Factor VIII (66).
Como hemos demostrado a lo largo de esta investigación, CD34 inmunomarca
no solo el endotelio sino también numerosos tumores de células
fusiformes, y como es de suponer, es utilizado para ayudar en el diagnóstico
de los tumores vasculares. En este trabajo examinamos 5 sarcomas de Kaposi (SK),
4 angiosarcomas (AS), 2 hemangioendoteliomas epitelioides (HEE), un hemagioendotelioma
(HE) y un hemangioma capilar lobular (HCL), usando CD31, Factor VIII, CD34,
Vimentina y Actina muscular específica.
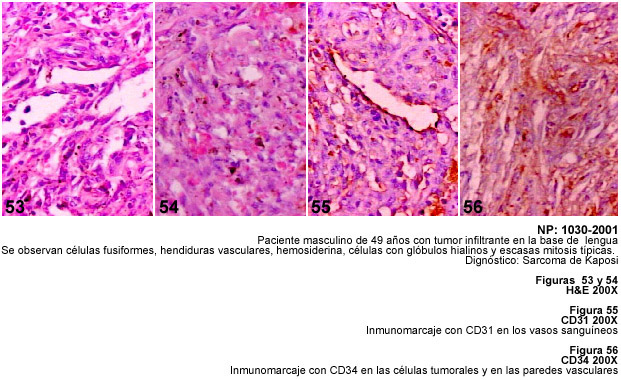
El sarcoma de Kaposi
(SK), es considerado en la reciente edición del Fascículo del
AFIP como una neoplasia vascular de malignidad intermedia (67). La discusión
sobre si el SK es una proliferación vascular estimulada por factores
exógenos o es una verdadera neoplasia, ha quedado saldada ante las evidencias
de monoclonalidad de sus células (68). Las diferencias entre los distintos
aspectos clínicos y patolígicos del SK han sido recientemente
publicadas en nuestro país a propósito de la Patología
del SIDA (69). Estos conceptos pueden resumirse en cuatro tipos de SK. El clásico:
en las extremidades inferiores de personas mayores, judíos Askenazis
o gentes del Mediterráneo. El SK del SIDA: diseminado en la piel, mucosas
y diferentes tejidos y órganos. El SK asociado a la inmunosupresión
iatrogénica: más común en pacientes transplantados. El
SK Africano: una forma endémica asociada a inmunosupresión crónica.
Sobre la relación entre el SK y los virus se han examinado muchos aspectos
y en la actualidad existe un virus, el Herpes Humano 8 (HHV 8), o virus del
SK, identificable en las células de SK y por lo tanto relacionado con
esta neoplasia (70,71). El diagnóstico diferencial del SK debe hacerse
con los angiosarcomas y hemangiomas y por su carácter de tumor fusocelular
hay que considerar los tumores descritos en este trabajo, en particular los
fibrohistiocíticos.
Los angiosarcomas
(AS) examinados en este trabajo correspondieron a la piel de cabeza y cuello
y de la región glútea y un AS del hígado. Esta neoplasia
es más común en las personas mayores de edad y puede mostrar diferentes
aspectos morfológicos con áreas papilares, fusocelulares, sólidas,
o formando canales anastomosados y puede simular un carcinoma indiferenciado,
o un melanoma (72). Se describe el AS postmastectomía (Síndrome
de Stewart-Treves). Más importante que el pleomorfismo y la apariencia
histológica en la evolución del AS, es su tamaño siendo
de mejor pronóstico los tumores menores de 5 cms (73). En general, con
CD31, Factor VIII, CD34 y Vimentina se corrobora el diagnóstico histológico
por la técnica de inmunohistoquímica.
El Hemangioendotelioma Epitelioide (HEE) fue descrito por Sharon Weiss y Franz Enzinger en 1982 como un tumor vascular que a menudo se confunde con carcinoma (74). Hace unos años publicamos una serie de casos que incluían un Hemangioma Histiocitoide para separar estas lesiones que con la enfermedad de Kimura (hiperplasia angiolinfoide con eosinofilia) son benignas, del HEE, un tumor vascular, con células de aspecto epitelioide, con frecuencia endovascular y mixoide considerado de grado intermedio de malignidad (75). En los tejidos blandos, el HEE puede ser confundido con un angiosarcoma epitelioide y con liposarcomas mixoides. Especialmente, puede confundirse con la variedad de células redondas y hasta con un sarcoma epitelioide, una neoplasia maligna en cuyo caso debe señalarse que estos tumores, usualmente en las extremidades y con aspecto multinodular, son positivas a CD34 casi e un 50% de los casos por lo que no ayuda este anticuerpo en su diagnóstico diferencial.
![]()
![]()